Problem:
The GC-content of a DNA string is given by the percentage of symbols in the string that are 'C' or 'G'. For example the string 'GCCGC" has a GC content of 100% but the string 'ATCGCAT' has a GC content of 37.5%.
Files with DNA string information in them are usually labelled using the FASTA format. In this format the string is introduced by a line starting off with '>', following with some labelling information. Subsequent lines contain the DNA string itself or the next string (see below).
Calculating GC content can be used as a tool to identify organisms, prokaryotes have GC content lower than 50% and eukaryotes' GC content hovers around 50%. If the DNA sample is long enough, we can identify species just by the GC content!
Given: At most 10 DNA strings in FASTA format
Return: The total number of rabbit pairs that will be present after n months, if we begin with 1 pair and in each generation, every pair of reproduction-age rabbits produces a litter of k rabbit pairs (instead of only 1 pair)
Sample Dataset:
>Rosalind_6404
CCTGCGGAAGATCGGCACTAGAATAGCCAGAACCGTTTCTCTGAGGCTTCCGGCCTTCCC TCCCACTAATAATTCTGAGG
>Rosalind_5959
CCATCGGTAGCGCATCCTTAGTCCAATTAAGTCCCTATCCAGGCGCTCCGCCGAAGGTCT ATATCCATTTGTCAGCAGACACGC
>Rosalind_0808
CCACCCTCGTGGTATGGCTAGGCATTCAGGAACCGGAGAACGCTTCAGACCAGCCCGGAC TGGGAACCTGCGGGCAGTAGGTGGAAT
Sample Output:
Rosalind_0808
60.919540
Solution:
First let's try opening the file in read mode and print out each line using the readlines() method so we can see what is inside the file. The readlines method for a file, essentially creates a list where everywhere line in the file is a list item.
For example, imagine the text file we have is:
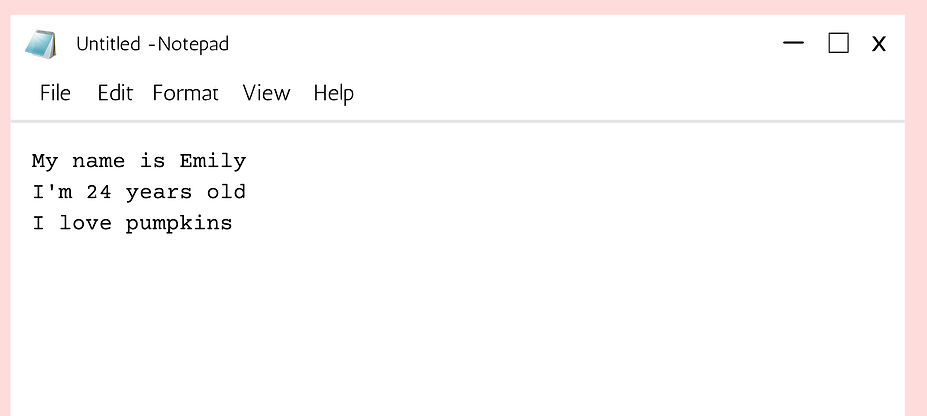_digital_art_x4.jpg)
We run a readlines code:
< >
file = open("emily.txt", "r")
print(file.readlines())
The output we get is:
["I'm Emily\n", 'I love oranges\n', "I'm 24"]
So each line becomes a list item. The \n you see denotes 'newline', so "My name is Emily\n" means there is a newline after that line. Which is true! But it is annoying to have that there, so we will need to use a strip() method to get rid of it.
The file we have is:

So let's write a function that breaks down our Rosalind file line by line into a list, with all the annoying \n s removed
< >
def read_file(file_path):
with open(file_path, "r") as file:
return [line.strip() for line in file.readlines()]
fasta_file = read_file("rosalind_gc.txt")
print(fasta_file)
We use list comprehension to strip away the \n and return a list of the lines in the Rosalind file. The output of the print is similar to what we've seen before:
['>Rosalind_6404', 'CCTGCGGAA...', '>Rosalind_5959', 'CCATCGGTA....']
Now that we've understood the basics of opening, stripping the lines and putting them into a list we can down to the refining the information.
We need to link every FASTA label (eg. Rosalind_5959) with it's related DNA string ('CCTGCGGAA...'). The listing of information in the file is sequential, that is: every label is directly succeeded by its DNA string and then a new label starts which is in turn succeeded by its DNA string (see file above).
And every label has an identifier, namely ">" so we can easily collate all the labels and their associated DNA strings in a dictionary and here is how we do it:
We first create an empty dictionary. Then write a for loop where every item in the fasta_file list is iterated over. If there is a ">" in the item, that means its a label (eg. Rosalind_5959) and we will create a variable label which is equal to item. If there isn't a ">" that means its a DNA string and it should be added to the previous label's item information.
< >
dict = {}
for line in fasta_file:
if ">" in line:
label = line
dict[label] = ""
else:
dict[label] += line
For example:
Imagine the first item in the list is ">Rosalind_6404". Since the item has a ">" identifier, it's a FASTA label and a variable label is created which is equal to ">Rosalind_6404". And then the label is made into a dictionary key. So our dictionary, if printed, would look like:
{'>Rosalind_6404', ''}
Now the next item in the list is "CCTGCGGAAGATCGGCACTAGAA..." - it's a mouthful! Since this item follows the >Rosalind_6404 label and doesn't have a ">", it means that the DNA string is of >Rosalind_6404 label as all information is sequential. So after an iteration over the second item, the dictionary would look like:
{'>Rosalind_6404': 'CCTGCGGAAGATCGGCACTAGAATAGCCAGAACCGTTTCTCTGAGGCTTCCGGCCTTCCCTCCCACTAATAATTCTGAGG'}
The next item is ">Rosalind_5959" and since it has a ">" identifier it will be made into a dictionary key too, so our dictionary at the end of this iteration will look like:
{'>Rosalind_6404': 'CCTGCGGAAGAT....',
'>Rosalind_5959': ''}
Now that we've made a dictionary with FASTA labels linked to their DNA strings, we can get to writing code for GC content calculation. This is a fairly simple mathematical operation:
number of C + number of G
total number of bases
GC Content =
So let's write a function that takes in a sequence of bases and spits out GC content to six decimal places:
< >
def gc_content(sequence):
return round(((sequence.count("C") + sequence.count("G")) / len(sequence)) * 100, 2)
So now we will calculate the GC content of each Rosalind label stored in our dictionary:
< >
def read_file(file_path):
with open(file_path, "r") as file:
return [line.strip() for line in file.readlines()]
fasta_file = read_file("rosalind_gc.txt")
print(fasta_file)
dict = {}
for line in fasta_file:
if ">" in line:
label = line
dict[label] = ""
else:
dict[label] += line
def gc_content(sequence):
return round(((sequence.count("C") + sequence.count("G")) / len(sequence)) * 100, 6)
result_dict = {}
for (key, value) in dict.items():
result_dict[key] = gc_content(value)
print(result_dict)
We create a results dictionary called result_dict() where we will store the dictionary key, the FASTA label, and it's corresponding GC content. By using dict.items() we access the key-value pairs of the dictionary and for every iteration of the key, the label, the GC content of it's corresponding DNA string is calculated and stored into the results dictionary as a new label-GC content pair. So the results dictionary will look like:
{'>Rosalind_6404': 53.75, '>Rosalind_5959': 53.57, '>Rosalind_0808': 60.91}
The hard part is over and now we simply need to pluck out which FASTA label has the largest GC value, and we will use Python's built-in max function:
< >
max_label = max(result_dict, key=result_dict.get)
print(max_label)
So what's going on here? The max function is fed the results dictionary as input, but what does key= result_dict.get do? This basically says that the code will iterate over all the key values in the dictionary, the FASTA labels in this case, and look at their corresponding GC content and then determine which GC content is the highest. After determining that, it returns the label value.
>Rosalind_0808
And now that we know the label with the highest GC content we can perform a simple dictionary get() method, and prettily format our answers!
< >
max_label = max(result_dict, key=result_dict.get)
print(f"{max_label[1:]}\n{result_dict.get(max_label)}")
It looks scary but is perfectly benign, trust me! The [1:] is to get rid of the ugly ">" identifier and the \n is to make a new line. The dictionary get method is used to get the GC content and none of this would be possible without a formatted string!
Output:
Rosalind_0808
60.91954
Copy and paste the output you get from running your code on the sample data onto the Rosalind answer terminal and submit. Annnd get yourself a cookie!